The Role of Phospho-ser-16-Phospholamban in Cardiac Physiology
Please rate these pages to support our infographic project:
Get free premium widgets for your blog and website.
Yael Lesin-Davis – University of Leeds
Cardiovascular disease is the single most prevalent cause of death in the United States and Europe. As such, research investigating the pathophysiology of the various forms of heart disease garners billions of dollars of research funding each year. An essential component of this research is focused on the molecular basis of normal heart function. Key to understanding these molecular mechanisms is phospholamban (PLN), an important regulatory protein that embeds within the membrane of the cardiomyocyte sarcoplasmic reticulum (SR). PLN plays a central role in calcium regulation and cardiac relaxation and as such, is a molecule that is deserving of attention and further understanding.
PLN binds to, and is key in regulating, the Ca2+ pump sarcoendoplasmic reticulum Ca2+ ATPase, SERCA2a. SERCA2a is responsible for removing cytosolic Ca2+ and transporting it into the SR. This function allows for cardiomyocyte relaxation (lusitropic effect). The ability of SERCA2a to sequester Ca2+ into the SR is inhibited by PLN, which binds to SERCA2a at resting Ca2+ levels. Phosphorylation of PLN relieves the inhibition of SERCA2a and enhances lusitropic effects and contractility. PLN can be phosphorylated at 3 residues: Serine-16, Threonine-17 and Ser-10. Polyclonal antibodies that are able to discriminate between phosphorylation of PLN at Ser-16 and phosphorylation at Thr-17 were first produced in 1994 [1]. Over the last 21 years, this discovery has facilitated cardiovascular research and progressed our understanding of PLN function. This article focuses on the intracellular pathways that regulate PLN phosphorylation at Ser-16.
PLN is phosphorylated on Ser-16 by cAMP, cGMP and peroxynitrite dependent pathways
cAMP-dependent protein kinase A (PKA) induced phosphorylation of PLN at Ser-16 has been observed in intact, perfused hearts when stimulated with catecholamines [2] [3]. Both β1- and β2- adrenergic receptors are coupled to Gs. The β1- adrenergic receptors stimulate adenylyl cyclase (AC), which manufactures cAMP (from ATP) stimulating the cAMP dependent PKA induced PLN phosphorylation. This relieves PLN inhibition of SERCA2a, increasing the calcium affinity of the SR Ca2+ pump by 2-3 fold. These processes make the PKA pathway the main regulator of cardiac relaxation [4] The Gs coupled β2-adrenergic receptor, however, does not augment PLN phosphorylation, indicating that this signal is compartmentalized [4][5].
The β2-adrenergic receptor also exists coupled to Gi, which inhibits AC, therefore suppressing the PKA pathway. Consequently, this inhibits PLN phosphorylation. This was shown with the use of inhibitors against Gi coupled adrenoceptors which augmented PLN-Ser-16 phosphorylation, suggesting that Gi as well as Gs control and the phosphatase: kinase equilibrium [6].
Additionally coupling to Gs are the 5-HT4 receptors. Activation of three 5HT4 receptor subtypes: 5 HT4a, 5HT4b and 5HT4c leads to a modest cAMP dependent phosphorylation of PLN at Ser-16. This pathway is similar to that of the β1-adrenoceptor [7].
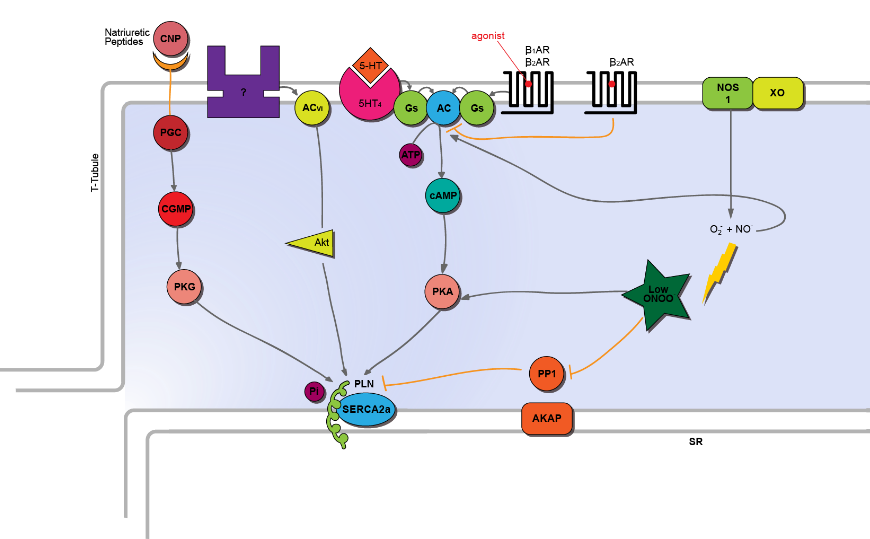
Cyclic GMP (cGMP) signalling also targets Ser-16 in cardiac myocytes. C-type Natriuretic Peptide (CNP) stimulates this cGMP pathway. CNP binding to its receptor, GC-B, stimulates coupled guanylyl cyclase, GC, which leads to an increase in cytosolic cGMP. This results in PLN phosphorylation at Ser-16 and subsequent disinhibition of SERCA2a [8].
NOS1 signalling increases PLN phosphorylation at Ser-16 via cGMP independent pathways. One of the pathways is cAMP dependent. NO activates AC, stimulating cAMP and subsequently PKA. This promotes the PLN phosphorylation at Ser-16 (as above) [9]. NOS1 can also couple to reactive signalling species, such as superoxide anion produced via xanthine oxidoreductase, to form peroxynitrite (ONOO-). Low levels of ONOO- enhance PLN- Ser-16 phosphorylation via promotion of the cAMP/PKA pathway and by inhibiting the phosphatases [9]. Additionally, low ONOO- phosphorylates PLN-16 via activating Akt; a protein which is also stimulated by AC type VI [10]. In contrast to low levels of ONOO-, high levels of ONOO- are detrimental to myocardial function. This is owing to its effect on decreasing cardiomyocyte contractility. The underlying mechanism of high ONOO- induced deleterious effects was found to be increased protein phosphatase 2A (PP2A) activity [11].
Counteracting kinase activity are the phosphatases. Protein phosphatases (PP): PP1, PP2A and PP2B (otherwise known as calcineurin) dephosphorylate PLN. PP1 is the main isotype of serine/ threonine phosphatase in cardiomyocytes, accounting for 90% of PLN dephosphorylation [12]. Inhibition of PP1, for example using cantharidin, increases PLN-Ser-16 phosphorylation and abbreviates relaxation time in the muscle. Other inhibitors of phosphatase activity have also been shown to augment Ser-16 phosphorylation of PLN and have been used in heart failure investigations. Various studies have shown increased phosphatase activity to exist in the failing heart. Inhibitor 1 (I-1) is an endogenous inhibitor of PP1 activity, which increases PLN phosphorylation. Adenoviral delivery of constitutively active inhibitor, I-1c, has been shown to decrease PP1 activity and preserve cardiac function in pigs with severe HF [13].
Sequestering phosphatase and kinase activity to the PLN are the protein kinase- A anchoring proteins (AKAPs). AKAPs are neither kinases nor phosphatases. However, they interact with and importantly anchor kinases at the SR, thereby conferring the spatial and temporal regulation that facilitates PLN phosphorylation [14]. One signal they compartmentalise is cAMP-PKA signal [15]. This occurs via the targeting of the PKA regulatory subunit, RII (which must be phosphorylated) to the AKAP helical region of AKAP: AKAP15/18δ. This sequesters PKA near its substrates, including PLN-Ser16, facilitating their phosphorylation [16].
In conclusion, Ser-16 phosphorylation is a rapid process that is facilitated by cAMP, cGMP and peroxynitrite pathways. This PLN-Ser-16 phosphorylation results in activation of SERCA2a and subsequent Ca2+ influx into the SR. This makes Ser-16 phosphorylation a major determinant of Ca2+ cycling & thus cardiac relaxation and contractility.
References
- Drago, G. A., & Colyer, J. (1994). Discrimination between two sites of phosphorylation on adjacent amino acids by phosphorylation site-specific antibodies to phospholamban. Journal of Biological Chemistry, 269(40), 25073–25077.
- LePeuch, C. J., LePeuch, D. A. M., Demaille, J. G. (1980). Phospholamban activation of the cardiac sarcoplasmic reticulum calcium pump. Physicochemical properties and diagonal purification. Biochemistry, 19, 3368-3373.
- Kranais, E. G. & Solaro, R. J. (1982). Phosphorylation of troponin I and Phospholamban during catecholamine stimulation of rabbit heart. Nature, 374, 135-141.
- Kuschel, M., Karczewski, P., Hempel, P., Schlegel, W.-P., Krause, E.-G., & Bartel, S. (1999). Ser16 prevails over Thr17 phospholamban phosphorylation in the beta -adrenergic regulation of cardiac relaxation. Am J Physiol Heart Circ Physiol, 276(5), H1625–1633. Retrieved from http://ajpheart.physiology.org/content/276/5/H1625
- Kuschel, M., Zhou, Y.-Y., Spurgeon, H. A., Bartel, S., Karczewski, P., Zhang, S.-J., … Xiao, R.-P. (1999). β2-Adrenergic cAMP Signaling Is Uncoupled From Phosphorylation of Cytoplasmic Proteins in Canine Heart. Circulation, 99(18), 2458–2465. doi:10.1161/01.CIR.99.18.2458
- Kuschel, M., Zhou, Y.-Y., Cheng, H., Zhang, S.-J., Chen, Y., Lakatta, E. G., & Xiao, R.-P. (1999). Gi Protein-mediated Functional Compartmentalization of Cardiac β2-Adrenergic Signaling. Journal of Biological Chemistry, 274(31), 22048–22052. doi:10.1074/jbc.274.31.22048
- Gergs, U., Baumann, M., Böckler, A., Buchwalow, I. B., Ebelt, H., Fabritz, L., … Neumann, J. (2010). Cardiac overexpression of the human 5-HT4 receptor in mice. American Journal of Physiology. Heart and Circulatory Physiology, 299(3), H788–98. doi:10.1152/ajpheart.00691.2009
- Pierkes, M., Gambaryan, S., Bokník, P., Lohmann, S. M., Schmitz, W., Potthast, R., … Kuhn, M. (2002). Increased effects of C-type natriuretic peptide on cardiac ventricular contractility and relaxation in guanylyl cyclase A-deficient mice. Cardiovascular Research, 53, 852–861. doi:10.1016/S0008-6363(01)00543-0
- Wang, H., Kohr, M. J., Traynham, C. J., Wheeler, D. G., Janssen, P. M. L., & Ziolo, M. T. (2008). Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. American Journal of Physiology. Cell Physiology, 294(6), C1566–75. doi:10.1152/ajpcell.00367.2007
- Gao, M. H., Tang, T., Guo, T., Miyanohara, A., Yajima, T., Pestonjamasp, K., … Hammond, H. K. (2008). Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. Journal of Biological Chemistry, 283(48), 33527–33535. doi:10.1074/jbc.M805825200
- Kohr, M. J., Davis, J. P., & Ziolo, M. T (2010). Peroxynitrite increases protein phosphatase activity and promotes the interaction of phospholamban with protein phosphatase 2a in the myocardium. NIH Public Access, 20(3), 217–221. doi:10.1016/j.niox.2009.01.003.
- Isoda, T., Paolocci, N., Haghighi, K., Wang, C., Wang, Y., Georgakopoulos, D., … Kass, D. a. (2003). Novel regulation of cardiac force-frequency relation by CREM (cAMP response element modulator). The FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 17, 144–151. doi:10.1096/fj.01-0981com
- Ishikawa K, Fish KM, Tilemann L, Rapti K, Aguero J, Santos-Gallego C, Lee A, Karakikies I, Xie C, Akar F, Shimada Y, Gwathmey JK, Samulski RJ, Sigg DC, Weber T, Kranias EG, Hajjar RJ (2014). Cardiac I-1c Over-expression with Reengineered AAV Improves Cardiac Function in Swine Ischemic Heart Failure. Mol Ther. 2014.
- Singh, P., Salih, M., & Tuana, B. S. (2009). Alpha-kinase anchoring protein alphaKAP interacts with SERCA2A to spatially position Ca2+/calmodulin-dependent protein kinase II and modulate phospholamban phosphorylation. The Journal of Biological Chemistry, 284(41), 28212–21. doi:10.1074/jbc.M109.044990
- McConnachie, G., Langeberg, L. K., & Scott, J. D. (2006). AKAP signaling complexes: getting to the heart of the matter. Trends in Molecular Medicine, 12(7), 317–23. doi:10.1016/j.molmed.2006.05.008
- Manni, S., Mauban, J. H., Ward, C. W., & Bond, M. (2008). Phosphorylation of the cAMP-dependent Protein Kinase (PKA) Regulatory Subunit Modulates PKA-AKAP Interaction, Substrate Phosphorylation, and Calcium Signaling in Cardiac Cells. The Journal of Biological Chemistry, 283(35), 24145–24154. doi:10.1074/jbc.M802278200v
Back to main infographic.
About us

Badrilla is a manufacturer of outstanding antibodies against cardiac proteins and phospho-proteins, and the manufacturer of leading kits for the study of protein S-palmitoylation.
Follow us on social
Contact us
We look forward to providing your favourite antibodies and kits
